
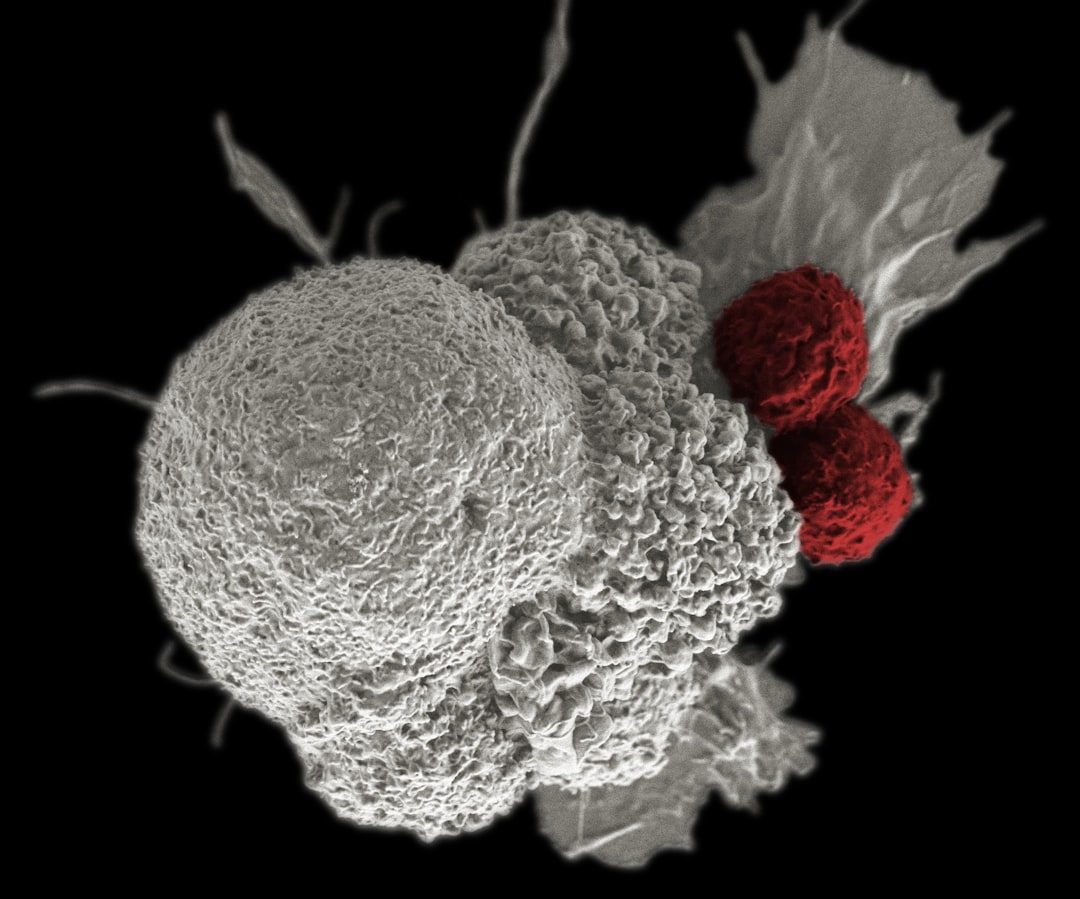



Kymriah and Yescarta CAR-T Approvals (2017)
The FDA approved the first two CAR-T cell therapies within two months—Kymriah for pediatric leukemia in August, Yescarta for lymphoma in October. Both were autologous therapies, custom-made from each patient's own T cells. The approvals created an entirely new regulatory category and reimbursement framework with no precedent. Cancer centers scrambled to build infrastructure for treatments costing $373,000-$475,000.
Established regulatory pathway for personalized cell therapies but exposed scalability and access challenges.
Six CAR-T therapies now approved; field shifted focus to allogeneic off-the-shelf approaches to solve manufacturing bottlenecks.
Tabelecleucel represents the next generation: allogeneic cells from healthy donors, manufactured in batches, potentially solving the supply and speed problems that plagued autologous CAR-T.